
MicroRNAs as Novel Regulators of Cardiac Arrhythmogenic Remodelling
In 1993 it was discovered that development of Caenorhabditis elegans is regulated by a short RNA fragment named Lin-4 that inhibits the expression of lin-14 by binding to the 3’ untranslated region of its mRNA.15,16 In the following years, these miRNAs were identified as key players in molecular signalling le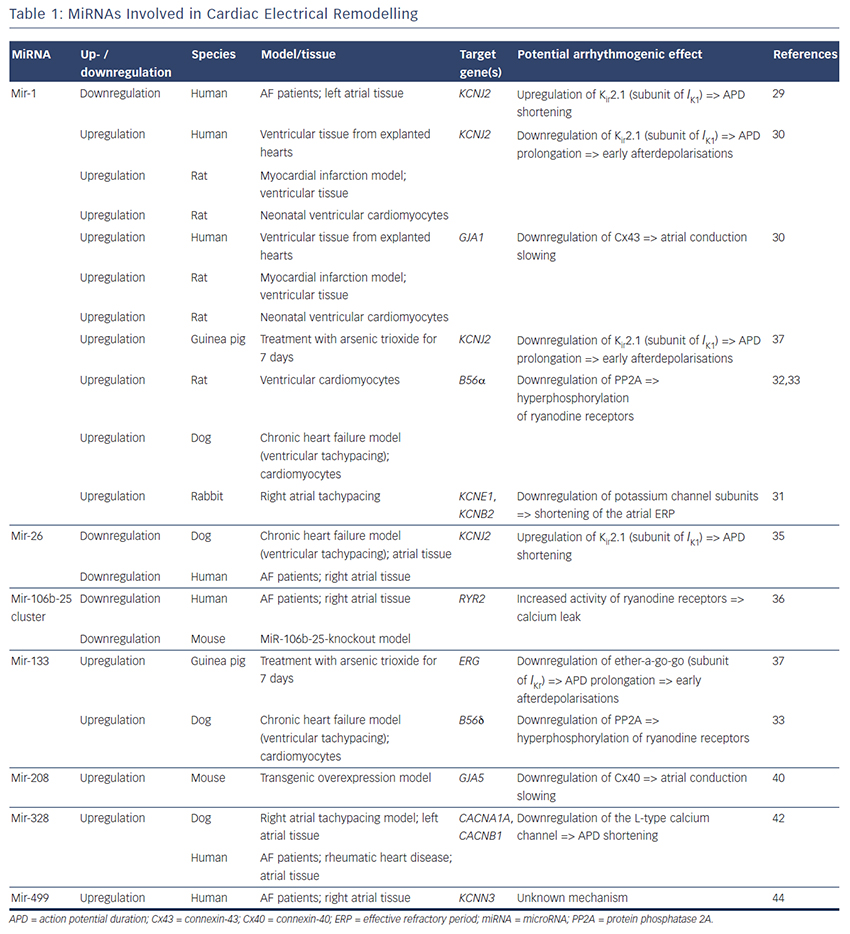 ading to disease. Although other noncoding RNA species have been discovered so far,17,18 we will focus on miRNAs in this review.
ading to disease. Although other noncoding RNA species have been discovered so far,17,18 we will focus on miRNAs in this review.
For a detailed description, especially of miRNA biogenesis we refer to previously published reviews.19–22 Briefly, miRNAs are short (approximately 22 base pairs), single-stranded, non-coding RNAs that regulate post-transcriptional gene expression. MiRNAs can induce down- and upregulation of genes either by direct inhibition of their target mRNA (causing direct downregulation of their target gene) or inhibition of an endogenous antagonist of another gene (causing indirect upregulation of a gene dependent on the miRNA’s target gene). MiRNAs are broadly conserved among species; to date, thousands of miRNAs have been discovered in plants, insects and mammals. According to the latest release of the online database miRBase (version 21.0, June 2014) 2,588 mature miRNAs have been identified in humans.23 Each miRNA can act on several target genes and each gene can be affected by several miRNAs. This complex regulatory network results in up- and downregulation of agonists and antagonists at the same time leading to the idea of miRNAs as fine tuners of gene expression.24
Experiments on genetic ablation of the enzyme dicer that is one of the key enzymes in miRNA biogenesis, revealed the crucial role for miRNAs in normal development as mice and zebrafish with dicer knockout are not viable.25 A cardiac-specific knockout of dicer in mice also resulted in premature lethality due to cardiac dilatation and heart failure.26 Finally, dicer is essential even in the adult organism as shown by da Costa Martins et al., who demonstrated that a reduced cardiac dicer activity leads to increased incidence of sudden cardiac death, cardiac hypertrophy and expressional switch from an adult to a fetal gene expression programme.27
These studies offered compelling evidence that microRNAs play an important role in normal cardiac development and disease.26,27 Therefore, a growing number of studies has been published evaluating the role of miRNAs in cardiac electrical and structural remodeling, potentially contributing to the initiation and maintenance of AF pathophysiology.7,9
miRNAs Involved in Electrical Remodelling
MiRNAs involved in cardiac electrical remodelling are miR-1, miR-26, miR-208a, miR-328 and miR-499 (Table 1, Figure 1). Their target genes are encoding ion channels, connexins or proteins involved in calcium signalling resulting in conduction slowing or shortening of the action potential duration, which are hallmarks of AF pathophysiology.
miR-1
MiR-1 and miR-133 are muscle-specific (skeletal and heart) miRNAs that are expressed from bicistronic clusters and, therefore, are among the most abundant miRNAs in the heart.28 Girmatsion et al. showed that miR-1 is downregulated in the heart of patients with persistent AF compared with patients with sinus rhythm.29 They demonstrated that this downregulation of miR-1 is accompanied by a significant upregulation of its target gene KCNJ2 that encodes Kir2.1, the subunit of the inward rectifier potassium channel IK1. Additionally, they showed that an ex vivo tachystimulation of atrial tissue resulted in downregulation of miR-1 and upregulation of Kir2.1 protein and IK1 density. An increased IK1 density is associated with a shortening of the APD and could therefore enable reentry and AF maintenance.
Yang et al. indicated another role for miR-1 in arrhythmogenesis as they confirmed KCNJ2 as an miR-1 target gene, but also identified GJA1 as an additional target gene that encodes connexin-43.30 They performed a miR-1 overexpression in an ischaemic rat model (which is in contrast to the miR-1 downregulation in patients with AF). Overexpressing miR-1 in the rat model led to downregulation of KCNJ2 and GJA1 resulting in APD prolongation and conduction slowing.
In 2013, Jia and colleagues published a study evaluating the role of miR-1 in rabbits.31 They performed right atrial tachypacing in rabbits for 1 week resulting in increased inducibility of AF due to a shortening of the atrial effective refractory period and an increase in the slowly activating delayed rectifier potassium current (IKs). They found an upregulation of miR-1 paralleled by a downregulation of KCNE1 (coding for the voltage-gated potassium channel subfamily E member 1, minK) and KCNB2 (coding for the voltage-gated potassium channel subfamily B member 2, Kv2.2). Using anti-miR-1 inhibitor oligonucleotides, they rescued the phenotype and prevented expressional changes of miR-1, KCNE1 or KCNB2.
Additionally, miR-1 is implicated in playing a role in calcium homeostasis. In rat cardiomyocytes, Terentyev et al. induced an overexpression of miR-1 resulting in  downregulation of its target B56α, a regulatory subunit of the protein phosphatase 2A (PP2A).32 PP2A now dissociates from the ryanodine receptor leading to hyperphosphorylation of this calcium channel. Hyperphosphorylated ryanodine receptors show arrhythmogenic spontaneous calcium release that can cause afterdepolarisations.32,33
downregulation of its target B56α, a regulatory subunit of the protein phosphatase 2A (PP2A).32 PP2A now dissociates from the ryanodine receptor leading to hyperphosphorylation of this calcium channel. Hyperphosphorylated ryanodine receptors show arrhythmogenic spontaneous calcium release that can cause afterdepolarisations.32,33
miR-26
MiR-26 has also been shown to regulate gene expression of KCNJ2.34,35 In a canine AF model and in human atrial samples, miR- 26 is downregulated while Kir2.1 is upregulated. Overexpression of miR-26 resulted in suppressed KCNJ2 expression, miR-26 knockdown resulted in enhanced KCNJ2 expression. These results were confirmed in a mouse model performing virus-induced miR-26 overexpression and miR-mask induced miR-26 knockdown:35 reduced/enhanced AF vulnerability was observed in miR-26 overexpression/knockdown.
miR-106b-25 cluster
Recently, Chiang and colleagues identified the miR-106b-25 cluster (miR-25, miR-93 and miR-106b) as mediators of electrical remodelling.36 They showed a downregulation in the atria of patients with AF and found an increased susceptibility for AF in miR-106b-25-knockout mice due to increased SR calcium release (SR calcium leak) mediated by an enhanced ryanodine receptor expression (which is the confirmed target gene of miR-93).
miR-133
Shan and colleagues evaluated the role of miR-133 on the QT interval in a guinea pig model.37 An upregulation of miR-133 resulted in significantly decreased protein levels of ERG, a subunit of the potassium channel responsible for the delayed rectifier potassium current Ikr, accompanied by a prolonged QTc interval and increased mortality rates. These findings were reproduced by direct application of miR-133 while miR-133 blockade using an antisense inhibitor abolished the effects. Interestingly, they also confirmed previous results of miR-1 in their model (upregulation of miR-1, downregulation of Kir2.1 protein).37
Matkovich et al. generated a miR-133 overexpressing mouse that displayed QT/APD prolongation and a reduced outward potassium current Ito,f.38 Although they could measure a reduced expression of the Ito,f subunit KCNIP2, they did not validate this gene as a miR-133 target.
Belevych et al. recently published a study in a canine model of heart failure.33 They demonstrated that miR-133 is upregulated leading to downregulation of its target gene B56δ, a catalytic subunit of PP2A with similar effects as B56α (that is downregulated as a target gene of miR-1, see above).
miR-208a
MiR-208 is an interesting miRNA as the two isoforms (miR-208a and miR-208b) are differentially expressed during cardiogenesis.39 MiR- 208a is encoded within an intron of the α-cardiac muscle myosin heavy chain gene MYH6 (adult isoform), whereas miR-208b is encoded within an intron of the β-cardiac muscle myosin heavy chain gene MYH7 (fetal isoform). MiR-208 isoforms are expressed along with their host genes: miR-208b is expressed during cardiogenesis while miR- 208a is expressed in adult hearts. Interestingly, pathological cardiac remodelling is associated with the induction of a fetal gene expression pattern with re-expression of MYH7 and, therefore, miR-208b.40,41 To evaluate miR-208 in cardiac remodeling, Callis et al. performed an overexpression of miR-208a in a mouse model and observed an increased vulnerability towards arrhythmias.40 They demonstrated that miR-208 targets GJA5 encoding the cardiac gap junction protein connexin-40 and therefore mediates pro-arrhythmogenic remodelling by altering gap junction expression.
miR-328
Another miRNA involved in calcium signalling is miR-328. It is upregulated in atrial tissue of patients with AF and in a canine AF model.42 CACNA1C and CACNB1, subunits of the L-type calcium channel, were identified as target genes of miR-328. In the canine model, an adenovirus-induced miR-328 overexpression caused reduced L-type calcium current and APD shortening and increased AF vulnerability. Knockdown of miR-328 with an antagomiR reversed these effects suggesting a potential role for miR-328 in cardiac electrical remodelling via the L-type calcium channel.
miR-499
KCNN3 encodes the calcium-activated potassium channel 3 (SK3) and is potentially involved in AF pathophysiology as common genetic variants within this gene are associated with AF.43 Recently, Ling and co-workers reported that miR-499 was elevated in human atrial tissue; SK3 protein expression, however, was downregulated.44 Finally, they provided evidence of a direct interaction between miR-499 and KCNN3 by in vitro overexpression and knockdown experiments. However, the authors analysed only eight patients (four with AF, four without AF [controls]) and did not report any enhanced arrhythmogenity in vitro or in vivo. Thus, the functional role of SK3 channels in AF pathophysiology still remains unclear as overexpression of SK3 channels in mice did not result in development of AF, but in an increased incidence of sudden death due to bradyarrhythmias and heart block.45