
Cardiac Energy Metabolism in Heart Failure
The heart is an organ with a high-energy demand. To perform continuous contractile activity, the myocardium hydrolyses more than 6 kg of adenosine triphosphate (ATP) daily.8 In normal conditions about 95 % of cardiac energy is obtained through the production of ATP from mitochondrial oxidative metabolism, w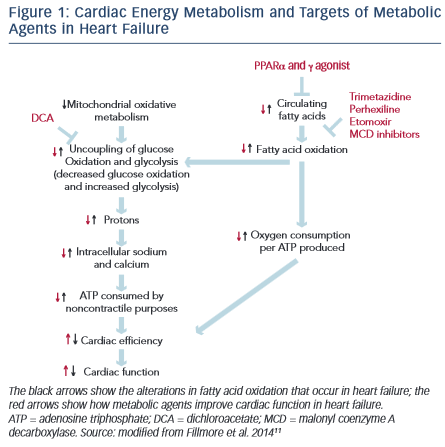 hile the remaining 5 % originates from glycolyticATP production.The changes in cardiac energy metabolism in heart failure are complex and depend on the stage and the cause of the heart failure.9 Mitochondrial oxidative metabolism is impaired in heart failure, resulting in a decrease in ATP and phosphocreatine levels in the failing heart. The decrease in glucose oxidation is more critical than changes in fatty acid oxidation.10 The proportion of ATP derived from mitochondrial fatty acid oxidation exceeds that derived from glucose oxidation, but more oxygen is required to produce ATP from fatty acid oxidation compared with glucose oxidation. In response to reduced oxidative metabolism the glucose uptake and glycolysis are elevated. High glycolysis rates and low glucose oxidation rates can result in an increase in the uncoupling of glycolysis from glucose oxidation, leading to the production of lactate and protons, which decreases the efficiency of the heart (see Figure 1).11
hile the remaining 5 % originates from glycolyticATP production.The changes in cardiac energy metabolism in heart failure are complex and depend on the stage and the cause of the heart failure.9 Mitochondrial oxidative metabolism is impaired in heart failure, resulting in a decrease in ATP and phosphocreatine levels in the failing heart. The decrease in glucose oxidation is more critical than changes in fatty acid oxidation.10 The proportion of ATP derived from mitochondrial fatty acid oxidation exceeds that derived from glucose oxidation, but more oxygen is required to produce ATP from fatty acid oxidation compared with glucose oxidation. In response to reduced oxidative metabolism the glucose uptake and glycolysis are elevated. High glycolysis rates and low glucose oxidation rates can result in an increase in the uncoupling of glycolysis from glucose oxidation, leading to the production of lactate and protons, which decreases the efficiency of the heart (see Figure 1).11
Recently, impaired mitochondrial oxidative metabolism in heart failure was defined using the term ‘metabolic remodelling’, as one component of a broader and more general concept of remodelling covering haemodynamic, neurohumoral, metabolic and inflammatory processes, causing changes in cardiomyocytes, endothelium, vascular smooth muscle cells as well as interstitial cells and matrix.12 This understanding of the processes of remodeling in heart failure has again drawn attention to the so-called metabolic vicious circle, which was proposed by Opie.13 The metabolic vicious circle includes the following sequence of events: the dilation of the myocardium in heart failure (A) leads to the adrenergic activation (B), that in turn hyperphosphorylates the sarcoplasmic reticulum (C) and increases the concentrations of circulating free fatty acids (D); free fatty acids inhibit mitochondrial function at the level of acyl carnitine transferase (E), thus inhibiting fatty acid oxidation and synthesis of ATP (F); plasma free fatty acids also inhibit pyruvate dehydrogenase (G) to promote anaerobic glycolysis (H), rather than oxidative metabolism (see Figure 2). This approach allows for consideration of therapies that target the cardiac metabolism as add-on therapy along with conventional treatment for heart failure.
Cardiac Energy Metabolism as a Potential Target for Therapy in Heart Failure
There is emerging evidence demonstrating that therapeutic regulation of cardiac metabolism by reducing fatty acid oxidation and/or increasing glucose oxidation may be an effective treatment for heart failure.11 Several pharmacological agents that modulate fatty acid metabolism by decreasing the supply of fatty acids to the heart, inhibiting fatty acid uptake and -oxidation or stimulating glucose oxidation have been developed (see Figure 1).

Both beta () blockers and nicotinic acid can decrease circulating free fatty acid levels and, therefore, indirectly reduce myocardial fatty acid oxidation and promote glucose use. blockers are fundamentally important in modifying the course of systolic heart failure.3 It is believed that inhibition of carnitine palmitoyl transferase-1 (CPT-1) activity, increased glucose oxidation and increased efficiency of oxygen use for ATP production may also be partially responsible for the beneficial effect of blockers in heart failure.14 Nicotinic acid, as a broad-spectrum lipid-regulating agent, reduces the frequency of cardiovascular disease events,15 but no additional benefit was found when used in conjunction with an intensive statin therapy.16 Unlike statins, there is no evidence that nicotinic acid reduces the incidence of heart failure in patients with coronary artery disease.
Peroxisome proliferator-activated receptor (PPAR) and PPAR agonists (fibrates and thiazolidinediones) also decrease the circulating free fatty acid supply to the heart, which results in reduced cardiac fatty acid oxidation rates. Fibrates lower the risk of major cardiovascular and coronary events compared with placebo, but do not affect the risk of cardiovascular or all-cause mortality.17 These lipid-regulating agents are not considered to be able to prevent the development of heart failure either. Thiazolidinediones are used to treat patients with type 2 diabetes; however, they may cause worsening of heart failure and increase the risk of heart failure-associated hospitalisation.18 Therefore, thiazolidinediones should not be used in patients with heart failure.3
Etomoxir and perhexiline, which are Inhibitors of CPT-1, decrease the activity of this rate-limiting enzyme for fatty acid -oxidation and thus limit fatty acid oxidation while favouring glucose oxidation (via glucose-fatty acid cycle, called the Randle Cycle).19 Etomoxir was initially developed as an antidiabetes agent and was then shown to improve left ventricular performance in animal studies.20,21 Etoximor was associated with improved exercise capacity but also increased liver transaminase levels in patients with heart failure.22 For this reason, etomoxir is not considered as a suitable modulator for use in heart failure. Perhexiline was initially developed as antianginal drug. Perhexiline inhibits the cardiac, but not the hepatic, isoform of CPT-1 and is associated with improved exercise capacity and left ventricular ejection fraction (LVEF) in patients with heart failure.23 In the late 1980s perhexiline was withdrawn worldwide, with exception of Australia and New Zealand where it remains licensed for the treatment of refractory angina.
Malonyl coenzyme A decarboxylase (MCD) inhibitors increase cardiac malonyl coenzyme A (CoA) levels, which inhibit CPT-1, thereby reducing mitochondrial fatty acid uptake. MCD inhibition leads to increased glucose oxidation, decreased fatty acid oxidation and improved insulin sensitivity.9 Pharmacological MCD inhibitors are under development for the treatment of myocardial ischaemia24 and obesity25. At present, no MCD inhibitors are available for clinical use.
Dichloroacetate (DCA) increases the activity of the mitochondrial pyruvate dehydrogenase complex by inhibiting pyruvate dehydrogenase kinase, and thereby increasing glucose oxidation. In an experimental study DCA was shown to reduce the rate of progression of left ventricular hypertrophy to heart failure.26 Currently, there is no clinical evidence that supports using DCA in heart failure.
Trimetazidine is a partial inhibitor of long-chain 3-ketoacyl CoA thiolase, the key enzyme in the -oxidation pathway. Trimetazidine shifts the myocardial energy metabolism from fatty acid -oxidation towards glucose oxidation, thereby increasing ATP generation and, ultimately, improving contractile function. Trimetazidine has been approved in more than 80 countries worldwide as an antianginal agent. Today, a vast amount of data confirm the efficacy of trimetazidine in heart failure. Ranolazine is similar in structure to trimetazidine and can inhibit fatty acid oxidation; however, this effect is much less pronounced.27 The main mechanism of action of ranolazine in myocytes is the inhibition of the late sodium current. Ranolazine is currently approved as an antianginal agent in USA and Europe. The impact of ranolazine on heart failure has only been investigated in a few clinical trials.28–30 Ranolazine has been shown to significantly increase left ventricular ejection fraction in patients with systolic and diastolic heart failure. The RanolazIne for the Treatment of Diastolic Heart Failure (RALI-DHF) study revealed that ranolazine improves measures of haemodynamics; however, there were no significant effects on relaxation parameters or N-terminal pro–B-type natriuretic peptide concentration in patients with heart failure with preserved ejection fraction.30 The value of this agent in the treatment of heart failure remains to be clarified.
The list of new therapies targeting cardiac metabolism is constantly expanding. Studies of mitochondria-targeted peptides (Szeto–Schiller peptides [especially SS-31] and coenzyme Q10), manganese superoxide dismutase mimetics, hormone replacement therapy, iron chelators and so on attract particular attention. Further experimental and clinical studies are required to confirm their efficacy in heart failure.