
Non-alcoholic Fatty Liver Disease and Cardiovascular Disease
Whereas the liver-related morbidity and mortality related to NAFLD/ NASH are well-documented and well-known, the consequences of NAFLD outside the setting of liver disease has long been unrecognised but gains growing attention. As already mentioned, NAFLD sometimes precedes the development of T2DM or the MetS, suggesting NAFLD is not simply a consequence but also a causal factor (and probably both) in their pathophysiology.5,17,18
Data are accumulating that patients affected by NAFLD have a higher risk of developing cardiovascular (CV) abnormalities, clinical CV events and even CV death.5,22 A first specific challenge in the interpretation of these data on the link between CVD and NAFLD is to distinguish between a timely correlation simply based on underlying risk factors that are shared by both conditions, or an independent contribution of NAFLD (after correction for these shared metabolic risk factors) in the subsequent development of CVD. The latter implies a specific pathophysiological contribution of the liver affected by NAFLD to the development of CV abnormalities. Elucidating the role of NAFLD in the development of CVD therefore constitutes a second challenge, in which, besides clinical data, studies in animal models might be helpful. Finally the question whether the role of NAFLD in the development of CVD is confined to NASH or is already present in NAFL needs to be answered. This question is particularly relevant for the treatment of NAFLD. If indeed the development of CVD is substantially influenced by NAFLD and NASH, its prevention might constitute an indication to treat NAFLD and its subtypes.
Clinical Data
The most convincing data on the role of NAFLD in CVD are those on the link between NAFLD and subclinical coronary heart disease (CHD). NAFLD, mostly diagnosed by ultrasound, has been shown to be an independent risk factor for the presence or future development of increased intima-media thickness, impaired flow-mediated vasodilatation, the presence of carotid atherosclerotic plaques, an increased coronary artery calcium score on cardiac computed 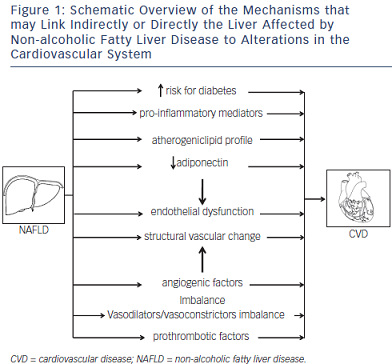 tomography and abnormal coronary flow reserve as a marker for impaired coronary microcirculation, both in cross-sectional and in follow-up studies, after correction for classical risk factors for CHD.23–36
tomography and abnormal coronary flow reserve as a marker for impaired coronary microcirculation, both in cross-sectional and in follow-up studies, after correction for classical risk factors for CHD.23–36
For clinical CHD, data are also emerging from large cohorts of patients, both cross-sectional and longitudinal studies, in community-based cohorts and in more selected patient groups (e.g. patients with T2DM, type 1 diabetes, patients undergoing coronary angiography or patients with documented NAFLD), that NAFLD is an independent predictor for clinical CHD, being the severity of the atherosclerotic lesions on coronarography or the occurrence of fatal and non-fatal CHD events.37–41 These data have been extensively reviewed elsewhere.5,6 Only a few studies did not confirm the independent relationship of NAFLD with incident CHD or showed it to be confined to patients with NAFLD who concomitantly met the diagnosis of the MetS.42,43 Overall the data strongly support the independent contribution of NAFLD to an increased risk of clinically relevant CHD, even after correction for an extended set of well-established risk factors for CHD.
Several studies also showed a link between NAFLD and alterations in cardiac metabolism,35,44 structure and haemodynamic function, such as myocardial insulin resistance and mitochondrial adenosine triphosphate (ATP) production, cardiac steatosis, myocardial hypertrophy and left ventricular diastolic dysfunction, not attributable to concomitant diabetes, obesity or arterial hypertension.44–50 The severity of these cardiac abnormalities correlated with the severity of the NAFLD. Finally NAFLD has been associated with an increased risk of autonomic dysfunction and cardiac arrhythmias (mainly atrial fibrillation).51–53 Interestingly, recent data have shown that NAFLD is also independently linked with QTc interval prolongation, a major risk factor for ventricular arrhythmias and sudden cardiac death, which might explain in part the increased CV mortality associated with NAFLD.54 Finally, congestive heart failure and aortic valve sclerosis have also been linked with NAFLD independently of known risk factors.55–57
Overall, although not all data are methodologically solid and most of the studies lack a diagnosis by the gold standard, the concept of NAFLD as being an independent contributor to the development of atherosclerosis and other functional and structural CV alterations, which subsequently lead to clinical CVD, seems sufficiently substantiated by the current evidence to integrate it in the clinical approach of both the NAFLD patient and the patient with CVD.
Pathophysiological Considerations
The mechanisms by which NAFLD influences the development of atherosclerosis and CVD is incompletely understood. NAFLD, T2DM, the MetS and CVD share many metabolic features and risk factors, leading to the concept that they belong to a complex multisystem disease with several organ manifestations and a complex interplay between the different entities, with multiple bidirectional cause–effect relationships. The specific contribution of one entity to the others is therefore difficult to discern, and there might be substantial inter-individual variability.
The contribution of NAFLD to CVD, seen as a unidirectional cause–effect relationship, can be either indirect or direct – the potential mechanisms are summarised in Figure 1. Firstly, as the liver is a key organ in both glucose and lipid homeostasis, it is not surprising that evidence is accumulating that NAFLD plays a role in the development of T2DM and the MetS, which are by themselves risk factors for CVD.5,17,18 This links NAFLD only indirectly to CVD.
NAFLD has indeed been shown to contribute to the development of T2DM. Several studies, mostly diagnosing NAFLD by ultrasound or liver enzymes, have shown that NAFLD precedes and predicts the future development of T2DM independent of obesity and other factors of the MetS.58,59 As insulin suppresses hepatic gluconeogenesis, NAFLD-associated hepatic insulin resistance results in mild hyperglycaemia, with a need for an increased insulin production to suppress hepatic glucose output and keep it within normal ranges. If Beta (β) cells cannot sustain this increased insulin secretion, patients develop impaired glucose tolerance and diabetes. Inflammatory mediators released by the inflamed liver in NASH might accelerate this process.60
Secondly, the liver might also contribute directly to the development of CVD. It is clear that NAFLD is associated with an atherogenic lipid profile.61 In NAFLD, production of triglyceride-rich very-low-density lipoprotein (VLDL) particles is increased.62 Insulin normally inhibits adipose tissue lipolysis (which is the main source of free fatty acids flux to the liver for incorporation in hepatic triglycerides) and hepatic VLDL secretion, both of which are hence increased in association with hepatic and adipose tissue insulin resistance.17,63 Subsequently, high-density lipoprotein cholesterol lowers and small dense low-density lipoprotein particles increase. Both conditions are highly atherogenic.
Endothelial dysfunction has been shown to be an early event in the development of atherosclerosis.64,65 Several studies have recently highlighted that insulin resistance at the endothelial level occurs early in the development of NAFLD and is already present after a few days of high-fat feeding, when steatosis develops but inflammation seems to still be absent.66–68 In the endothelium, insulin stimulates nitric oxide (NO) release leading to vasodilatation, and an impairment of insulin signaling leads to a reduced vasodilatory response to acetylcholine (ACh), which is used as a well-established hallmark of endothelial dysfunction.68 Steatosis leads to impaired endothelial NO synthase (eNOS) phosphorylation and hence impaired NO response to insulin, contributing to an increase in intrahepatic resistance.66 Conversely, insulin-sensitizing drugs improve endothelial function in NAFLD, as demonstrated by an improvement in the vasodilatory response to ACh.66 These changes occur early in the development of NAFLD, before the development of inflammation and before peripheral insulin resistance can be documented.68 Although the exact mechanisms need to be further elucidated, these findings point towards a pivotal role of steatosis in the impairment of endothelial function as a primary event preceding extrahepatic events.
The increased intrahepatic resistance is not only attributable to endothelial dysfunction based on reduced NO production because of endothelial cell insulin resistance. An imbalance in locally produced vasodilators and vasoconstrictors has also been documented.68 Steatosis was shown to be associated with a disturbed production of endothelin 1 and of cyclooxygenase-mediated vasoactive prostaglandins. Although data in humans are scarce and mainly restricted to the measurement of metabolites of vasoactive substances in peripheral blood (both in cirrhosis and in NAFLD patients), alterations have been documented and reflect potential systemic effects of what happens inside a liver affected by NAFLD, and hence its contribution to the development of CV alterations.69
Furthermore, steatosis also induces structural abnormalities of liver vasculature that also contributes to the associated increase in intrahepatic resistance.68 The pathophysiology of these structural alterations is currently unknown. Angiogenic factors have been shown to play a role in the intrahepatic vascular changes in cirrhosis and are also studied in NASH.70 Altered levels of angiogenic factors (vascular endothelial growth factor and its soluble receptors 1 and 2) have also been documented in the peripheral blood of patients with NASH.69 This is used as an argument to support the hypothesis of the role of angiogenic factors in the pathophysiology of NASH, but it also might help us to understand the link between the liver and CVD. Although this has not been proven so far, it can be hypothesized that the altered concentrations of angiogenic factors exert their effects in the extrahepatic vascular beds. The role of angiogenic factors, which not only influence vascular growth but also have vasoactive properties in the pathogenesis of atherosclerosis, has been well-documented.71
Prothrombotic factors have also shown to play a role in the progression of liver disease.72 Several metabolic risk factors are prothrombotic,73 but the role of the liver in this prothrombotic state has been poorly documented. Increased levels of prothrombotic factors have been described in patients with NASH.74 Although the liver is the main source of most of these coagulation factors, the causal role of the liver has not been proven. We studied an extensive panel of coagulation factors in a large series of histologically proven NAFLD patients, showing that mainly plasminogen activator inhibitor 1 (PAI-1) is increased in association with NASH, whereas some of the other factors (e.g. factor VIII, protein S) are elevated in relation with metabolic parameters, as was shown previously73,75 but the alterations of the latter do not correlate with liver histology.74 Furthermore, PAI-1 was also elevated in relation to the severity of liver histology. Together these findings point towards an independent contribution of NAFLD severity to a prothrombotic state that might contribute to CVD.
Adiponectin is another factor that might represent a link between NAFLD and CVD. Adiponectin, secreted by adipocytes, is decreased in obesity76,77 but also in NAFLD in relation to the histological severity of NAFLD after correction for body mass index (BMI).78,79 Adiponectin has insulin-sensitizing, anti-inflammatory and anti-atherogenic properties80 and directly affects endothelial function by eNOS messenger RNA (mRNA) stabilisation and eNOS phosphorylation.81 Furthermore, adiponectin stimulates circulating angiogenic cells.82 NAFLD-associated adiponectin decrease might therefore contribute to the development of CVD.
Inflammatory mediators can also contribute to the increased risk of CVD. NASH is associated with an increased intrahepatic production of pro-inflammatory cytokines, which are also increased systemically.83–85 One of the cytokines that are increased in NASH is Interleukin 6 (IL6), which stimulates angiotensin II in vascular smooth muscle cells with an associated production of reactive oxygen species, which in turn interact with NO production and activity.69 Other inflammatory mediators released by the liver might also contribute to atherogenesis.
Although all these mechanisms are plausible links between the liver affected by NAFLD and the development of CVD, no studies to date have scientifically proven to really represent a cause–effect relationship. Several mechanisms are most probably concomitantly present, and might substantially differ between patients. Further study is hence needed to gain mechanistic insight into the pathophysiology of the NAFLD–CVD axis, with an individualised approach, both preventive and therapeutic, as the ultimate goal.