
Aetiology and Classification of Sleep-disordered Breathing in Heart Failure
SDB describes a range of conditions in which there is an abnormality of the breathing pattern during sleep. By definition, a cessation of oro-nasal airflow for over 10 seconds is termed an apnoea, while a reduction in airflow amplitude by 30 % or more for 10 seconds associated with a desaturation of 3 % or more is a hypopnoea.9 Events are further classified into obstructive, central or mixed depending on the presence, absence or characteristics of thoraco-abdominal movement during the episode. The number of events per hour of sleep is the apnoea–hypopnoea index (AHI). Up to 5 events per hour is considered normal, 5–15/hour mild, 15–30/hour moderate and over 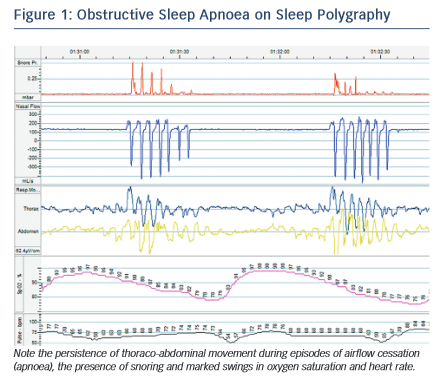 30 events/hour severe SDB.9
30 events/hour severe SDB.9
Although OSA and CSA have different underlying mechanisms, they are both driven by the internal pathophysiological environment found in HF. In OSA, the interruption of airflow is caused by collapse of the airway at the pharynx during inspiration. Pharyngeal muscle tone is decreased during sleep and, in HF, rostral shift of fluid from oedematous extremities to the pharynx may exacerbate this tendency. Research has demonstrated a significant increase in neck circumference overnight associated with a decrease in leg circumference in those with HF and OSA.10 Obesity and retrognaithism also predispose to this condition, although patients with HF and OSA are more frequently non- obese compared with patients with normal cardiac function.11 OSA is characterised by abrupt cessation of breathing as the airway occludes, accompanied by respiratory muscle effort and often snoring, followed by compensatory hyperventilation to restore arterial partial pressure of carbon dioxide (PaCO2) (see Figure 1).
In CSA, the abnormal breathing pattern is mediated by dysregulation in the respiratory centres of the brainstem. While the mechanisms of CSA are not fully understood and may vary between individuals, the following processes are likely to contribute. In normal physiology, depth and frequency of breathing (and hence minute ventilation) is driven by PaCO2 sensed by the chemoreceptors in the carotid bodies, aortic arch and brainstem. Small rises in PaCO2 result in a transient increase in ventilation that restores the PaCO2 to within the narrow normal physiological range. In patients with CSA, there is an exaggerated hypercapnic ventilatory response, so that small rises in PaCO2 result in inappropriate hyperventilation, which drives the PaCO2 down. If the PaCO2 falls to below the ‘apnoeic threshold’ the neural drive becomes insufficient to stimulate a breath and an apnoea occurs. Lesser reductions in neural drive result in hypopnoea. The PaCO2 subsequently rises again and the cycle repeats.12 Exaggerated sympathetic nervous activity in HF is thought to underlie this excessive response and patients with CSA and HF have a low resting PaCO2.5 In addition, prolonged circulation time in HF results in a lag between the PaCO2 sensed in the brainstem and that at the alveoli, which exacerbates an overshoot of the feedback loop. Oedema and congestion in the lungs, which may progress during the night, stimulates pulmonary J receptors resulting in a reflex hyperventilation. A characteristic crescendo-decrescendo pattern of breathing in CSA is termed ‘Cheyne-Stokes respiration’ (see Figure 2).