
Epidemiology
PO is the second most common clinical presentation of AHF syndromes (AHFS), though the prevalence and in-hospital mortality may exhibit substantial geographic variation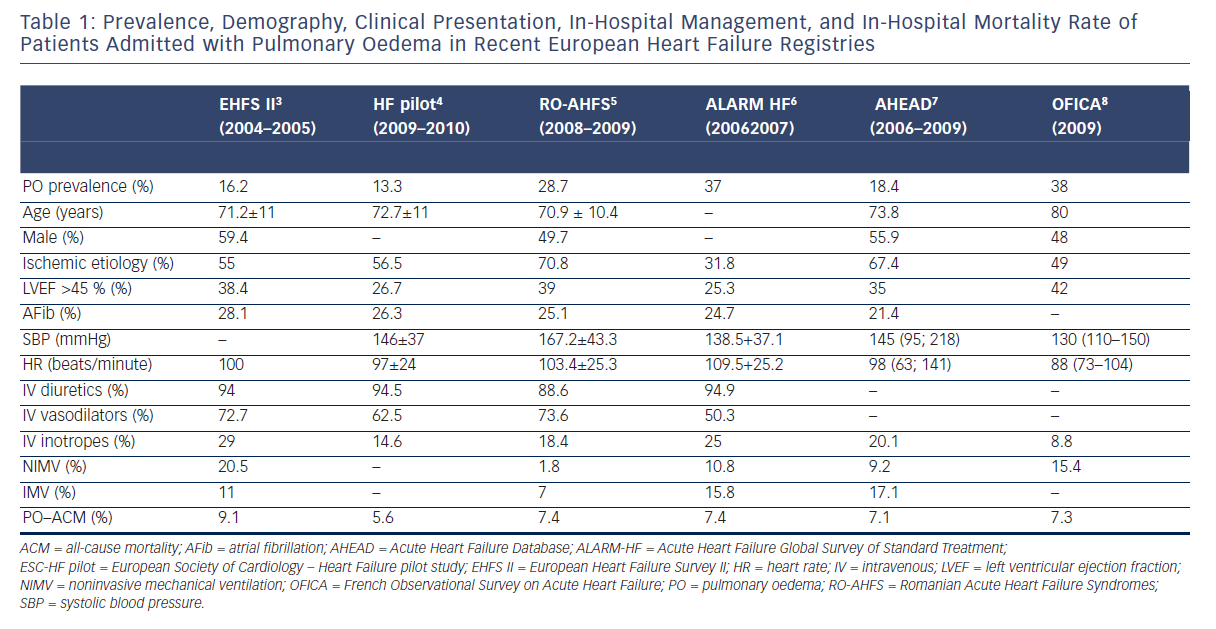 (see Table 1).3–10 Moreover, overlapping features among AHF clinical profiles and/or the confounding presence of noncardiac comorbidities may lead to incomplete or inaccurate classification. These patients are often not included in clinical trials due to their disease severity and inability to wait for study medication to be initiated. PO may be a deadly clinical manifestation of HF (see Table 1) and recent experience11 suggests the vast majority of deaths occurred soon after admission.
(see Table 1).3–10 Moreover, overlapping features among AHF clinical profiles and/or the confounding presence of noncardiac comorbidities may lead to incomplete or inaccurate classification. These patients are often not included in clinical trials due to their disease severity and inability to wait for study medication to be initiated. PO may be a deadly clinical manifestation of HF (see Table 1) and recent experience11 suggests the vast majority of deaths occurred soon after admission.
Pathophysiologic Mechanisms
The pathogenesis of hydrostatic PO has been attributed predominantly to a difference in Starling forces12,13 (i.e. fluid extravasation attributable to an increased hydrostatic or reduced oncotic pressure gradient across the intact alveolo-capillary barrier). Furthermore, capacity of the lymphatic system to remove fluid from the interstitial space and to drain into the systemic veins is dependent on systemic venous pressure and the integrity of the lymphatics. The clinical picture of PO is dominated by pulmonary congestion due to an acutely increased afterload. 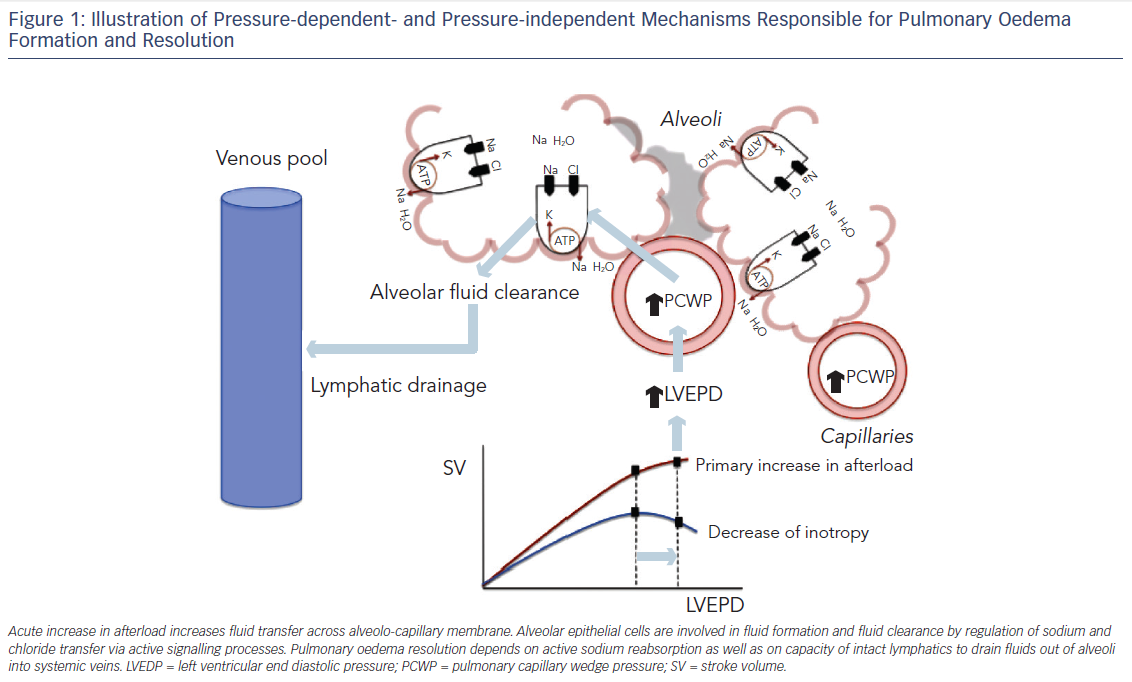 Increases in afterload can be the primary mechanism responsible of PO inducing acute cardiac dysfunction and pulmonary congestion, or can be secondary to large cardiac dysfunction (see Figure 1).
Increases in afterload can be the primary mechanism responsible of PO inducing acute cardiac dysfunction and pulmonary congestion, or can be secondary to large cardiac dysfunction (see Figure 1).
In a recent study,14 the mechanism of PO was described as a consequence of acutely increased afterload in patients with decreased systolic and diastolic capacity to adapt to changes in loading in the presence of maintained right ventricular function. It has been hypothesized that PO patients respond to increased afterload with an increase in heart rate (HR) and peripheral vasoconstriction rather than an acute adaptation in cardiac dimensions.11 This suggests there are smaller cardiac dimensions and a higher HR and diastolic blood pressure (DBP) in PO patients compared with other profiles.11
Peripheral congestion and body weight increases are documented in a relatively small proportion of PO patients, 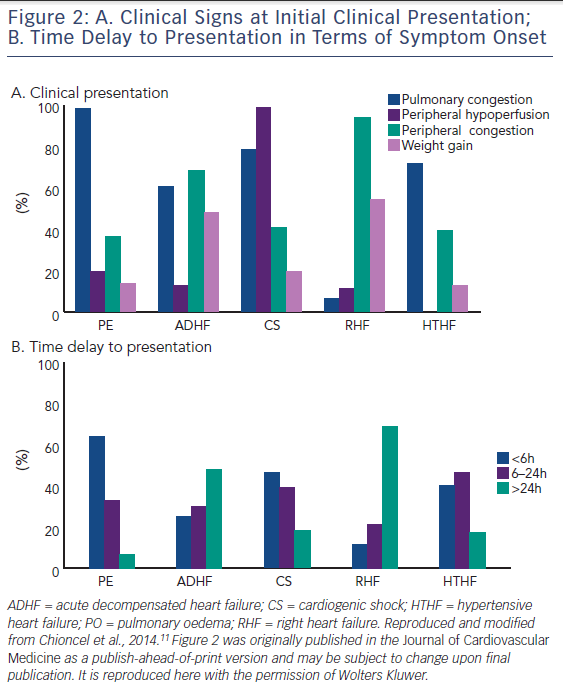 suggesting that relative volume redistribution as opposed to an absolute increase in total body fluid may play a major role in the pathophysiology of the PO phenotype (see Figure 2). The concept of fluid redistribution in PO is further supported by the studies, which have documented that the majority
suggesting that relative volume redistribution as opposed to an absolute increase in total body fluid may play a major role in the pathophysiology of the PO phenotype (see Figure 2). The concept of fluid redistribution in PO is further supported by the studies, which have documented that the majority
of patients gain little or no weight before heart failure hospitalization, despite the fact that filling pressures rise significantly.15–17 Sympathetically stimulated reduction in venous capacitance acts to shift volume out of the splanchnic vessels and increase the effective circulating blood volume,18 leading to increases in preload in the absence of any changes in total body weight.
Although pressure-related mechanisms were considered sufficient to explain PO, recent studies show that cardiogenic PO is critically regulated by active signalling processes (see Figure 1), suggesting that endothelial and alveolar responses may contribute critically to the formation of hydrostatic PO. Absorption of excess alveolar fluid is an active process (see Figure 1) that involves transport of sodium out of alveolar air spaces with water following the sodium osmotic gradient. Active transepithelial transport of sodium from the airspaces to the lung interstitial space is a primary mechanism driving alveolar fluid clearance. This mechanism depends on sodium uptake by amiloridesensitive sodium channels on the apical membrane of alveolar type II cells followed by extrusion of sodium on the basolateral surface by the Na-K-ATPase.19–22
A major part of cardiogenic PO formation results from active epithelial secretion of chlorine and secondary fluid flux into the alveolar space.23 Transepithelial chlorine secretion is triggered by inhibition of epithelial sodium uptake and mediated via cystic fibrosis transmembrane conductance regulator (CFTR) and Na-K-2Cl-co-transporter 1 (NKCC1).23
Active sodium transport across the alveolar epithelium is also regulated via basolateral Na-K-ATPase and acute elevation of left atrial pressure inhibits active sodium transport by decreasing the number of Na-K-ATPase at the basolateral membrane of alveolar epithelial cells.22 Regulation of the Na-K-ATPase is elicited by stimulation of dopaminergic,24 β2 adrenergic receptors,25,26 and aldosterone, and inhibited by oabain-like compounds.22,24–26 Furthermore, recent data suggest that lung injury and barrier dysfunction may play a role in the formation and resolution of congestion and pulmonary oedema.26