
Pathophysiology of Heart Failure
Blood pressure (BP) is defined as the product of cardiac output (C.O.) and systemic vascular resistance (SVR), where C.O. is the product of stroke 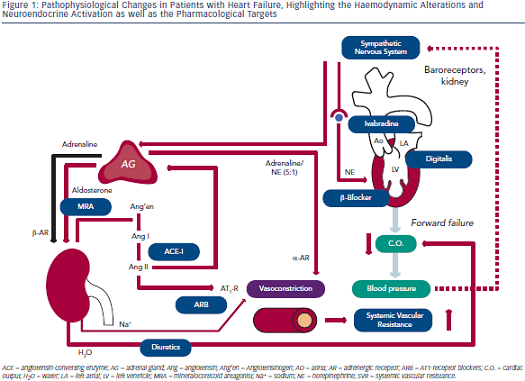 volume (SV) and heart rate (HR). Systolic dysfunction of the left ventricle (LV) reduces SV and thus C.O., which results in decreased BP. A decrease in BP is sensed by baroreceptors in the carotid artery and the aorta, which activates the sympathetic nervous system that is centrally controlled in the brain stem (medulla oblongata), triggering increased release of norepinephrine in the myocardium and of epinephrine from the adrenal glands to the bloodstream (see Figure 1). While epinephrine increases SVR via vascular alpha (α)-adrenergic receptors, norepinephrine has positive inotropic and chronotropic effects in the heart by stimulating beta (β)-adrenergic receptors, increasing HR and SV. Together, sympathetic activation increases BP and therefore has beneficial effects on short-term haemodynamics while in the long run, leads to LV remodeling and dysfunction7 through activating pro-hypertrophic signaling pathways8 and inducing apoptosis.9,10 Furthermore, elevated heart rate per se is associated with adverse prognosis in patients with HF.11
volume (SV) and heart rate (HR). Systolic dysfunction of the left ventricle (LV) reduces SV and thus C.O., which results in decreased BP. A decrease in BP is sensed by baroreceptors in the carotid artery and the aorta, which activates the sympathetic nervous system that is centrally controlled in the brain stem (medulla oblongata), triggering increased release of norepinephrine in the myocardium and of epinephrine from the adrenal glands to the bloodstream (see Figure 1). While epinephrine increases SVR via vascular alpha (α)-adrenergic receptors, norepinephrine has positive inotropic and chronotropic effects in the heart by stimulating beta (β)-adrenergic receptors, increasing HR and SV. Together, sympathetic activation increases BP and therefore has beneficial effects on short-term haemodynamics while in the long run, leads to LV remodeling and dysfunction7 through activating pro-hypertrophic signaling pathways8 and inducing apoptosis.9,10 Furthermore, elevated heart rate per se is associated with adverse prognosis in patients with HF.11
Activation of renal sympathetic efferent nerves to the kidney with subsequent activation of β-adrenergic receptors as well as reduced renal blood flow increase the release of renin from the kidney, which converts angiotensinogen to angiotensin I, which is then converted to angiotensin II (Ang II) by the angiotensin-converting enzyme (ACE;see Figure 1). Ang II induces vasoconstriction via type 1 Ang II (AT1) receptors and stimulates the release of aldosterone from the adrenal glands. Aldosterone, in turn, increases sodium (Na+) and water (H2O) retention in the kidney, which elevates intravascular volume and hence, SV and BP. Similar to the sympathetic nervous system, the activation of the renin-angiotensin-aldosterone system (RAAS) has beneficial short-term effects on BP, but adverse long-term effects on LV remodeling and prognosis, which is largely related to the activation of pro-hypertrophic and maladaptive signaling pathways in the heart.12
With progressive LV remodeling and dysfunction, the filling pressure of the LV increases, which triggers the production and release of brain natriuretic peptide (BNP) from LV myocardium, with the N-terminal pro-BNP being used as the currently most sensitive and reliable biomarker for the haemodynamic status and prognosis of patients with HF. Through its natriuretic action, BNP tends to antagonise the effects of RAAS activation. As a consequence of elevated LV filling pressures, left atrial pressure increases, inducing atrial dilation and fibrosis and thus, providing a substrate for atrial fibrillation (AF).13 Furthermore, elevated LV and atrial filling pressures trigger pulmonary congestion, which is the basis for the leading symptom of HF (i.e. dyspnoea). When pulmonary congestion continues to increase pulmonary artery pressures, the right heart is also exposed to elevated afterload and thus, after a prolonged time, right ventricular (RV) decompensation may lead to the development of oedema in arms, legs and potentially the gut.
Considering these pathophysiological changes during HF, the treatment of these patients primarily aims to antagonise neuroendocrine activation and congestion (see Figure 1), which in concert trigger maladaptive remodeling of the LV.12
Christoph Maack is supported by the Deutsche Forschungsgemeinschaft (Heisenberg Programm, SFB 894) and the Deutsche Herzstiftung (Margret Elisabeth Strauß Projektförderung).