
Association of Genotype with Clinical Characteristics
Studies of the association of genotype with clinical ARVD/C phenotype are challenging due to broad clinical variabili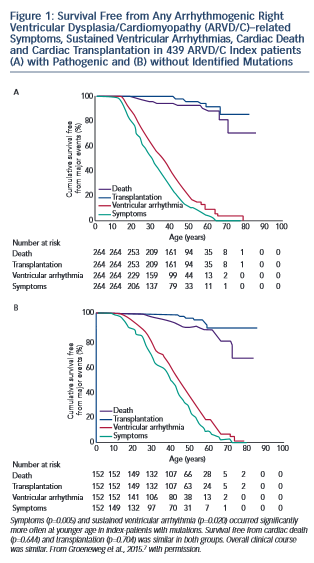 ty even within families who share both genotype and environment. However, clinically useful patterns of genotype/phenotype associations have begun to emerge. Broadly, the clinical course of ARVD/C does not differ substantially between ARVD/C index patients with and without identifiable mutations.7 As shown in Figure 1, regardless of mutation status, ARVD/C is a highly arrhythmic condition. Most index patients experience symptoms and sustained ventricular arrhythmias, but only a minority heart failure or transplant. The single exception to this pattern of overall similarity was that index patients with mutations had a significantly younger age of symptom onset and first ventricular arrhythmias than patients without mutations. This pattern of earlier onset is commonly seen when comparing strongly genetic forms of diseases with those with complex multifactorial inheritance.
ty even within families who share both genotype and environment. However, clinically useful patterns of genotype/phenotype associations have begun to emerge. Broadly, the clinical course of ARVD/C does not differ substantially between ARVD/C index patients with and without identifiable mutations.7 As shown in Figure 1, regardless of mutation status, ARVD/C is a highly arrhythmic condition. Most index patients experience symptoms and sustained ventricular arrhythmias, but only a minority heart failure or transplant. The single exception to this pattern of overall similarity was that index patients with mutations had a significantly younger age of symptom onset and first ventricular arrhythmias than patients without mutations. This pattern of earlier onset is commonly seen when comparing strongly genetic forms of diseases with those with complex multifactorial inheritance.
In a pattern also observed among many genetic diseases, several groups of investigators have found that ARVD/C patients with more than one desmosomal mutation have worse phenotypes than patients with a single or no mutation. Rigato et al. observed increased penetrance and worse arrhythmic outcomes in 21 Italian carriers of multiple mutations in comparison to 113 carriers of single-desmosomal mutations.43 They showed that being a carrier of more than one mutation was the most significant risk factor for malignant arrhythmic events and sudden death in their population. In another study, a higher incidence of cardiogenic syncopal events occurred in multiple mutations carriers among a Chinese population of ARVD/C patients.27 Finally, in an analysis of 577 desmosomal, PLN and TMEM43 mutation carriers drawn from the Johns Hopkins and Dutch ARVD/C Registries, the 4 % of patients with multiple mutations had significantly earlier occurrence of sustained ventricular tachycardia/ventricular fibrillation, a lower rate of survival free from sustained ventricular arrhythmias and more frequent left ventricular (LV) dysfunction, Class C heart failure and cardiac transplantation.23 Taken together, these data suggest an effect of gene dosage on ARVD/C phenotype. Based on these data, more aggressive management of carriers of multiple mutations is indicated.
There are also several clinically relevant associations between mutations in specific desmosomal genes and ARVD/C characteristics. A higher prevalence of LV involvement occurs among carriers of DSP mutations. This association was initially recognised by Norman et al. among a British population of ARVD/C patients in 2005.44 Defects in the C-terminal of the protein were associated with early and predominant involvement of the LV and a high occurrence of sudden death.45 In a large study by Bhonsale et al.,23 DSP carriers experienced more than fourfold more LV dysfunction (40 %) and Class C heart failure (13 %) than their large group of PKP2 carriers. LV involvement is also more likely among cases with PLN mutations. (Most PLN carriers have the Dutch founder mutation c.40_42delAGA that has been identified in 13 % of ARVD/C index patients in the Netherlands).1 The TMEM43 S358L founder mutation most prevalent in Newfoundland is associated with very high disease penetrance and arrhythmic risk among male carriers in comparison to other ARVD/C patients.46
Incomplete Penetrance and Variable Expressivity
While improved understanding of the genetic basis of ARVD/C has been helpful in diagnosing and managing patients, familial ARVD/C remains a clinically heterogeneous disorder characterised by incomplete age-related penetrance and significant variable expressivity.47 With the expansion of genetic testing for ARVD/C and integration of genetic test results into the diagnostic criteria,42 clinicians are increasingly confronted with caring for at-risk mutation carriers. While the literature reports up to 83 % penetrance of desmosmal mutations (in Spanish carriers of a DSP c1339C>T mutation),48 this likely is an overestimate, or at least a ceiling. Families initially ascertained and enrolled in genetic research will have higher than typical penetrance as this is what makes them attractive for genetic studies. In the large report by Bhonsale et al. of over 500 desmosomal mutation carriers,23 roughly one-third met ARVD/C 2010 Diagnostic Task Force Criteria (TFC). When family members are diagnosed, they often have a milder course than probands.7 While the full explanation for this phenotypic heterogenity remains unclear, there is increasing evidence that exercise plays a major role in disease penetrance and arrhythmic risk.
Cynthia A James holds grants sponsored by the National Society of Genetic Counselors and the Barth Syndrome Foundation. The Johns Hopkins ARVD/C Program is supported by the Dr Francis P Chiaramonte Private Foundation, the Leyla Erkan Family Fund for ARVD Research, the Dr Satish Rupal and Robin Shah ARVD Fund at Johns Hopkins, the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J Harrison Family, the Peter French Memorial Foundation and the Wilmerding Endowments.