
Mechanisms of Catecholamine Toxicity
It is evident that an abnormal response to a catecholamine surge leads to the development of TTC, but the mechanisms by which it does so have not been clearly established (see Figure 2). Introducing rats to a stressful situation such as immobilisation induces the typical apical ballooning pattern of TTC.12 It has been shown that pre-treatment with a beta-blocker, alpha-blocker or a combination of the two prevented the occurrence of TTC in immobilised rats.12 Furthermore, giving rats an alpha or 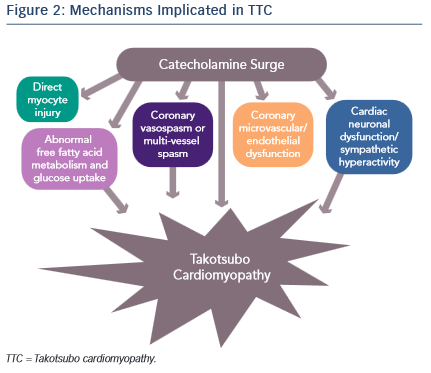 beta agonist actually induced TTC.13 Plasma catecholamine levels in patients that have undergone a major emotional or physical stressor with subsequent TTC are found to be at supraphysiological levels 1–2 days after the initial stressor and half the peak values after 1 week.14 These levels are twice as high as are observed in patients with Killip class III MI.14 There is evidence of increased sympathetic activity in patients with TTC as confirmed by 123I-meta-iodobenzylguanidine (123I-mIBG) single-photon emission computed tomography (SPECT) imaging.15 Additionally, the plasma levels of catecholamine precursors and neuronal as well as extraneuronal breakdown products are elevated, implicating increased catecholamine synthesis as well as neuronal and extraneuronal metabolism.14
beta agonist actually induced TTC.13 Plasma catecholamine levels in patients that have undergone a major emotional or physical stressor with subsequent TTC are found to be at supraphysiological levels 1–2 days after the initial stressor and half the peak values after 1 week.14 These levels are twice as high as are observed in patients with Killip class III MI.14 There is evidence of increased sympathetic activity in patients with TTC as confirmed by 123I-meta-iodobenzylguanidine (123I-mIBG) single-photon emission computed tomography (SPECT) imaging.15 Additionally, the plasma levels of catecholamine precursors and neuronal as well as extraneuronal breakdown products are elevated, implicating increased catecholamine synthesis as well as neuronal and extraneuronal metabolism.14
Epicardial coronary artery spasm causing ischaemia has also been implicated in TTC. The elevation of catecholamine levels stimulates the α1 receptors on the coronary vasculature leading to coronary vasoconstriction with resultant ischaemia. This is supported by ST elevations being a common finding in TTC despite the absence of obstructive coronary artery disease. However, the ST elevations in TTC are more diffuse and do not typically follow a distinct epicardial distribution. Thus, it is proposed that multi-vessel coronary vasospasm may be responsible for the development of TTC. Tsuchihashi et al. demonstrated that 21 % (n=48) of patients have inducible vasospasm with use of acetylcholine.16 Another systematic review showed 27.6 % of subjects have provoked coronary vasospasm in response to acetylcholine or ergovine.17 Thus, while an important contributor towards the development of TTC, multi-vessel epicardial coronary spasm does not explain the majority of TTC cases.
Alternatively, coronary microvascular and endothelial dysfunction is another hypothesised mechanism of TTC. One of the major arguments for this mechanism is that electrocardiogram (ECG) changes are diffuse and not suggestive of a single coronary artery territory and on cardiac catheterisation the majority of patients do not have obstructive coronary artery disease. Additionally, the peak troponin level and the extent of ECG changes in patients with TTC are shown to correlate to the severity of endothelial dysfunction.18 These patients are found to have a reduced coronary blood flow during the acute TTC event that remains impaired after the resolution of TTC left ventricular dysfunction, suggesting that coronary microvascular dysfunction is present at baseline in patients with TTC.19,20 Moreover, female patients with a history of TTC have evidence of impaired microvascular dilation in response to acetylcholine as well as a significantly lower increase in peak coronary blood flow compared with age-matched women with normal microvascular responses.20
The reactive hyperaemia peripheral arterial tonometry (PAT) is a reflection of changes in peripheral endothelial function and correlates with coronary endothelial function. TTC patients compared with post-MI and post-menopausal control women demonstrate more adverse mental stress PAT scores compared with the other groups, consistent with a persistently abnormal physiological response in TTC patients.21 Excessive vasoconstriction and impaired endothelium dependent vasodilation is suggested to cause this abnormal response to mental stress testing.20
It has previously been proposed that left ventricular outflow tract (LVOT) obstruction leads to TTC. The theory is that catecholamine release causes LOVT obstruction thus increasing the mechanical stress on the cardiac apex and subsequently leads to myocardial stunning.22 A recent study reveals that dobutamine causes mid-ventricular outflow gradients in both patients with and without a history of TTC.22 This means that there are no differences in patients with or without a TTC history that would predispose them to develop TTC. It is not likely that LVOT obstruction is the sole cause of TTC given that the incidence of LVOT obstruction in TTC is about 20–25 %.23
A fourth proposed mechanism is that the elevated catecholamine levels may cause direct myocyte injury. In previous studies it is shown that norepinephrine causes an increase in cyclic adenosine monophosphate (cAMP)-mediated intracellular calcium overload that is responsible for myocyte toxicity.24 Furthermore, catecholamines can be a source of oxygen-derived free radicals that then interfere with intracellular calcium transporters, leading to excess calcium influx and subsequent myocyte damage.14,25 One way in which increased intracellular calcium may cause cell injury is through the high-energy phosphate deficiency that results from excessive activation of the calcium dependent ATPase, therefore leading to impaired mitochondrial function.24 Histologically, myocytes have been shown to have contraction band necrosis and mononuclear inflammation, both characteristic signs of catecholamine toxicity that differ from ischaemic-induced necrosis.25 Contraction band necrosis and mononuclear inflammation are also seen in other catecholamine-excess states, such as subarachnoid haemorrhage and pheochromocytoma.25 It has been hypothesised that the fibrosis seen in TTC does not reach a critical value due to the concomitant release of anti-fibrotic factors by norepinephrine, and is the reason that TTC is reversible.25
Another theory of acute-myocardial stunning via alterations in cell signalling is proposed as a further cause of the direct effect of catecholamines on the heart. Normally, norepinephrine binds to the β1-adrenoreceptors that are coupled to Gs proteins, subsequently increasing cAMP to activate protein kinase A and therefore cause increased contractile response via release of calcium. Epinephrine has the same effect but with a higher affinity for the β2-adrenoreceptors, which also leads to a positive inotropic response. At supraphysiological epinephrine levels, the β2-adrenoreceptors couple to Gi protein instead of the Gs protein therefore leading to a negative inotropic effect on the heart.2 Additionally, although there are more sympathetic nerve endings in the basal myocardium, there is a higher concentration of β-adrenoreceptors at the apex thus explaining the typical apical ballooning pattern due to apical akinesis.26 Once the epinephrine levels return to normal, the β2-adrenoreceptors are once again coupled to the Gs protein.2 The switch to Gi protein may have a protective role in the myocardium. High levels of β1-adrenoreceptor-mediated Gs activation can stimulate apoptotic pathways therefore switching to Gi protein via β2-adrenoreceptors inhibits apoptosis from occurring.27 Additionally, as previously mentioned, intracellular calcium overload by the Gs receptor can be cardiotoxic, so the switch to the Gi pathway may be cardioprotective.
Neurogenic stunned myocardium is another mechanism proposed to cause TTC. The fact that an emotional stressor causes TTC suggests that the brain plays a role in inducing cardiac injury. Patients with subarachnoid haemorrhage are found to have elevated catecholamine levels. These patients, particularly females, demonstrate similar findings as those in TTC such as diffuse ST elevation without obstructive coronary artery disease on cardiac catheterisation, significantly reduced wall motion of the apex, and histological changes consistent with those of catecholamine excess, such as contraction band necrosis.28,29 In subarachnoid haemorrhage, ischaemia of the hypothalamus releases excess amounts of norepinephrine and subsequently leads to myocyte stunning.28 A more recent study with three patients shows direct evidence that brain activation is implicated in TTC by measuring cerebral blood flow via SPECT.30 Cerebral blood flow was increased in the hippocampus, brainstem and basal ganglia and decreased in the pre-frontal cortex during the acute phase of TTC in all three subjects.30 This pattern of brain activation is is found in acute stress and with sympathetic activation.30
In a recent study by Vaccaro et al., TTC patients are found to have increased SNS activity at baseline and a decreased baroreceptor response compared with congestive heart failure (CHF) patient controls.31 CHF patients have elevated SNS activity, thus this study demonstrated that TTC patients have even higher sympathetic activity. Moreover, TTC patients also had decreased baroreceptor inhibition of the SNS. In this study, SNS activity was directly measured by assessing muscle sympathetic nerve activity (MSNA).31 The study only determined an association between increased SNS and decreased baroreceptor response but did not determine causality. Also, by looking at the musculature, the study focused on the SNS activation of the peripheral vasculature but not the heart itself. However, it does suggest that autonomic dysfunction could play a role in TTC. This can further be supported by the fact that many patients with haemorrhage or ischaemic stroke have high occurrence of TTC, and may serve as an alternative explanation for neurogenic stunning.